Ирелми (Wainua) инструкция по применению
- 📜 Инструкция по применению Ирелми
- 💊 Состав препарата Ирелми
- ✅ Применение препарата Ирелми
- 📅 Условия хранения Ирелми
- ⏳ Срок годности Ирелми

Владелец регистрационного удостоверения:
Лекарственная форма
|
|
Ирелми |
Раствор для п/к введения 45 мг: шприц-ручка 0.8 мл
рег. №: ЛП-(009384)-(РГ-RU)
от 24.03.25
- Действующее
|
Форма выпуска, упаковка и состав препарата Ирелми
Раствор для п/к введения прозрачный, от бесцветного до желтого цвета.
| 1 мл | 1 шприц-ручка (0.8 мл) | |
| эплонтерсен (в форме эплонтерсена натрия) | 56 мг | 45 мг |
Вспомогательные вещества: натрия дигидрофосфата дигидрат, динатрия фосфат, натрия хлорид, хлористоводородная кислота концентрированная1, натрия гидроксид1, вода д/и.
1 Добавляется в виде разбавленного раствора в воду для инъекций для доведения рН до 7.4.
0.8 мл - шприц-ручки* (1) - пачки картонные с контролем первого вскрытия.
* первичная упаковка: по 0.8 мл в шприц стеклянный типа I с иглой, защитным колпачком для иглы и ограничителем хода поршня; шприц встроен в шприц-ручку, состоящую из блока привода и блока для шприца.
Фармакологическое действие
| Данный лекарственный препарат подлежит дополнительному мониторингу. Это позволит быстро выявить новую информацию по безопасности. Просьба к работникам системы здравоохранения сообщать о любых подозреваемых нежелательных реакциях. Порядок сообщения о нежелательных реакциях представлен в разделе "Побочное действие". |
Механизм действия
Эплонтерсен представляет собой конъюгированный с N-ацетилгалактозамином 2`-O-2-метоксиэтил (2`-MOE)-модифицированный химерный гапмерный антисмысловой олигонуклеотид (ASO) со смешанной основой из фосфоротионированных (PS) и фосфодиэфирных (PO) межнуклеотидных связей. Конъюгат N-ацетилгалактозамина обеспечивает направленную доставку ASO в гепатоциты. Селективное связывание эплонтерсена с матричной РНК транстиретина (мРНК TTR) в гепатоцитах приводит к деградации как мутантной, так и нормальной (дикого типа) мРНК TTR. Это препятствует синтезу белка TTR в печени, что приводит к значимому снижению уровня мутированного белка TTR и белка TTR дикого типа, секретируемого печенью в кровоток.
TTR является белком-носителем для ретинол-связывающего белка 4 (RBP4), который является основным переносчиком витамина А (ретинола). Таким образом, ожидается, что снижение концентрации TTR в плазме крови приведет к снижению концентрации ретинола в плазме крови до уровня ниже нижней границы нормы.
Фармакодинамические эффекты
В клиническом исследовании у пациентов с ATTRv-PN, получавших эплонтерсен, снижение концентрации TTR в сыворотке крови отмечали уже при первой оценке (5-я неделя), и концентрация TTR продолжала снижаться до 35-й недели. Устойчивое снижение концентрации TTR было отмечено на протяжении всего периода лечения (85 недель). Среднее значение (стандартное отклонение (СО)) процентного снижения уровня TTR в сыворотке крови по сравнению с исходным уровнем составило 82.1% (11.7) на 35-й неделе, 83.0% (10.4) на 65-й неделе и 81.8% (13.4) на 85-й неделе при лечении эплонтерсеном. Сходное снижение концентрации TTR в сыворотке крови по сравнению с плацебо было отмечено независимо от пола, расы, возраста, региона, массы тела, статуса кардиомиопатии, предшествующего лечения, статуса мутации Val30Met, стадии заболевания и исходного наличия клинического диагноза семейной амилоидной кардиомиопатии (FAC) (рисунки 1а и б).
Рисунок 1. Форест-диаграмма различия между группами лечения в среднем НК для изменения уровня TTR (г/л) в процентах относительно исходного уровня в основных подгруппах (исследование NEURO-TTRansform) (популяция полного анализа)
а) на 35-й неделе
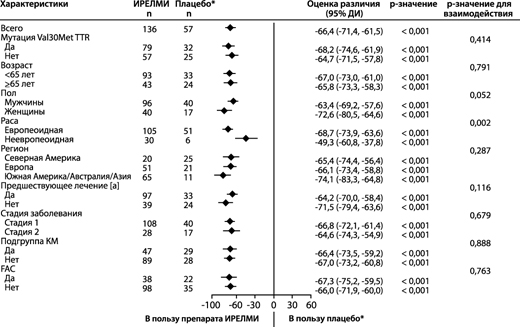
* Группа с внешним плацебо из другого рандомизированного контролируемого исследования (NEURO-TTR).
[а] Ранее получавшие лечение тафамидисом или дифлунисалом.
На основе MMRM, скорректированной с помощью показателей предрасположенности с категориальными эффектами для лечения, времени, взаимосвязи между лечением и временем, а также стадии заболевания, мутации Val30Met, предшествующего лечения и фиксированных ковариат для исходного уровня и взаимосвязи между исходным уровнем и временем.
Модели подгрупп также включали взаимосвязи между лечением и подгруппой, временем и подгруппой и лечением, временем и подгруппой. В промежуточный анализ на 35-й неделе включены только данные до 35-й недели.
В подгруппу КМ вошли пациенты с диагнозом FAC на момент включения в исследование или исходной толщиной межжелудочковой перегородки ≥13 мм при отсутствии артериальной гипертензии [в анамнезе или диагностированной во время исследования].
Представлено различие между группами лечения в отношении средних НК на 35-й неделе (Ирелми - плацебо) с 95% ДИ (нескорректированное). ДИ - доверительный интервал; среднее НК – среднее значение, рассчитанное по методу наименьших квадратов; MMRM – модель смешанных эффектов с повторными измерениями; TTR – транстиретин, КМ – кардиомиопатия, FAC – семейная амилоидная кардиомиопатия.
б) на 65-й неделе
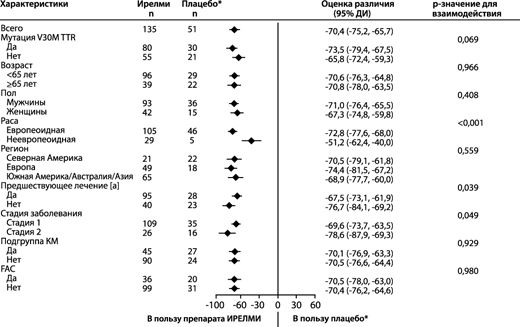
* Группа с внешним плацебо из другого рандомизированного контролируемого исследования (NEURO-TTR).
[а] Ранее получавшие лечение тафамидисом или дифлунисалом.
На основе MMRM, скорректированной с помощью показателей предрасположенности с категориальными эффектами для лечения, времени, взаимосвязи между лечением и временем, а также стадии заболевания, мутации Val30Met, предшествующего лечения и фиксированных ковариат для исходного уровня и взаимосвязи между исходным уровнем и временем.
Модели подгрупп также включали взаимосвязи между лечением и подгруппой, временем и подгруппой и лечением, временем и подгруппой.
В подгруппу КМ вошли пациенты с диагнозом FAC на момент включения в исследование или исходной толщиной межжелудочковой перегородки ≥13 мм при отсутствии артериальной гипертензии (в анамнезе или диагностированной во время исследования).
Представлено различие между группами лечения с точки зрения средних, полученных методом НК на 65-й неделе (Ирелми – плацебо) с 95% ДИ (нескорректированное). ДИ - доверительный интервал; среднее НК – среднее значение, рассчитанное по методу наименьших квадратов; MMRM – модель смешанных эффектов с повторными измерениями; TTR – транстиретин, КМ – кардиомиопатия, FAC – семейная амилоидная кардиомиопатия.
Электрофизиология сердца
Формальное исследование длительности интервала QT, скорректированной по частоте сердечных сокращений (QTc), при применении препарата Ирелми не проводили. Возможность удлинения интервала QTc при применении эплонтерсена оценивали в рандомизированном плацебо-контролируемом исследовании с участием здоровых добровольцев. При дозе, в 2.7 раза превышающей рекомендуемую дозу 45 мг эплонтерсена, клинически значимого влияния на интервал QT не отмечали.
Иммуногенность
В клиническом исследовании с участием пациентов с ATTRv-PN после 84-недельного периода лечения (медиана продолжительности лечения 561 день (80 недель), диапазон от 57 до 582 дней) у 58 пациентов (40.3%) были выявлены антитела к лекарственному препарату в связи с применением препарата.
У пациентов с наличием антител к эплонтерсену не было выявлено клинически значимого влияния на эффективность, безопасность, фармакокинетику и фармакодинамику препарата Ирелми.
Клиническая эффективность и безопасность
Эффективность и безопасность препарата Ирелми оценивали в рандомизированном многоцентровом открытом исследовании с внешним плацебо (NEURO-TTRansform), в которое были включены 168 пациентов с ATTRv-PN. Пациенты были рандомизированы в соотношении 6:1 для получения препарата Ирелми 45 мг посредством подкожного введения каждые 4 недели (N=144) или инотерсена 284 мг 1 раз в неделю (N=24) в качестве референтной группы. Из 144 пациентов, рандомизированных для получения эплонтерсена, 140 (97.2%) пациентов продолжали лечение до 35-й недели, 135 (93.8%) - до 65-й недели и 130 (90.3%) – до 85-й недели.
Внешняя группа плацебо состояла из когорты пациентов, получавших плацебо в опорном исследовании инотерсена: рандомизированное двойное слепое плацебо-контролируемое многоцентровое клиническое исследование с участием взрослых пациентов с ATTRv-PN (NEURO-TTR). Эта когорта получала подкожные инъекции плацебо 1 раз в неделю. В обоих исследованиях использовали идентичные критерии включения пациентов в исследование.
Характеристики групп эплонтерсена и внешнего плацебо, как правило, являлись сходными, а потенциальные нарушения баланса в основных исходных характеристиках (возраст, статус мутации Val30Met, стадия заболевания и предшествующее лечение) были учтены в предварительно определенном статистическом анализе. Исходные демографические характеристики и характеристики заболевания представлены в таблице 1.
Таблица 1. Исходные демографические характеристики и характеристики заболевания в исследовании NEURO-TTRansform (популяция для оценки безопасности)
| Плацебо* (N=60) | Ирелми (N=144) | |
| Возраст, лет | ||
| Среднее значение (СО) | 59.5 (14.1) | 53.0 (15.0) |
| Медиана (мин., макс.) | 63 (28, 81) | 51.5 (24, 82) |
| <65, n (%) | 34 (56.7) | 100 (69.4) |
| 65-74, n (%) | 17 (28.3) | 36 (25.0) |
| ≥75, n (%) | 9 (15.0) | 8 (5.6) |
| Мужской пол, n (%) | 41 (68.3) | 100 (69.4) |
| Раса, n (%) | ||
| Монголоидная | 3 (5.0) | 22 (15.4) |
| Негроидная или афроамериканцы | 1 (1.7) | 5 (3.5) |
| Европеоидная | 53 (88.3) | 112 (78.3) |
| Другое | 2 (3.3) | 3 (2.1) |
| Смешанная | 1 (1.7) | 1 (0.7) |
| Этническая принадлежность, n (%) | ||
| m | 60 | 142 |
| Испанское или латиноамериканское происхождение | 7 (11.7) | 22 (15.5) |
| Предшествующее лечение тафамидисом или дифлунисалом, n (%) | ||
| Да | 36 (60.0) | 100 (69.4) |
| Стадия заболевания ATTRv-PN1, n (%) | ||
| Стадия 1 | 42 (70.0) | 115 (79.9) |
| Стадия 2 | 18 (30.0) | 29 (20.1) |
| Комбинированный балл по mNIS+7, среднее значение (СО) | 74.8 (39.0) | 81.3 (43.4) |
| Суммарный балл по Норфолк QoL-DN | ||
| m | 59 | 137 |
| Среднее значение (СО) | 48.7 (26.8) | 44.1 (26.6) |
| Мутация Val30Met TTR, n (%) | ||
| Да2 | 33 (55.0) | 85 (59.0) |
| Нет3 | 27 (45.0) | 59 (41.0) |
| Glu89Gln, Glu109Gln | 0 | 1 (0.7) |
| Leu58His, Leu78His | 3 (5.0) | 4 (2.8) |
| Phe64Leu, Phe84Leu | 3 (5.0) | 5 (3.5) |
| Ser50Arg, Ser70Arg | 1 (1.7) | 2 (1.4) |
| Ser77Tyr, Ser97Tyr, S97Y | 5 (8.3) | 3 (2.1) |
| Thr49Ala, Thr69Ala | 0 | 1 (0.7) |
| Thr60Ala, Thr80Ala | 8 (13.3) | 4 (2.8) |
| Val122Ile, Val142Ile | 1 (1.7) | 4 (2.8) |
| Другое3 | 6 (10.0) | 35 (24.3) |
| Классификация NYHA, n (%) | ||
| I | 40 (66.7) | 105 (72.9) |
| II | 20 (33.3) | 39 (27.1) |
| Продолжительность заболевания с момента постановки диагноза ATTRv-PN (месяцы), среднее значение (СО) | 39.3 (40.3) | 46.8 (58.1) |
| Продолжительность от начала симптомов ATTRv-PN (месяцы), среднее значение (СО) | 64.0 (52.3) | 67.7 (50.9) |
| Диагноз семейной амилоидной кардиомиопатии (FAC)4, n (%) | 22 (36.7) | 39 (27.1) |
| Критерии, использованные для подтверждения клинического диагноза FAC4, n (%)5 | ||
| Биопсия сердца | 5 (22.7) | 1 (2.6) |
| Результат эхокардиографии | 17 (77.3) | 24 (61.5) |
| Другое | 0 | 24 (61.5) |
| Продолжительность заболевания с момента постановки клинического диагноза FAC4 из ИРК (месяцы), среднее значение (СО) | 21.0 (22.5) | 18.5 (21.4) |
| Продолжительность от начала симптомов FAC4 (месяцы), среднее значение (СО) | 34.1 (29.3) | 36.3 (63.8) |
| NT-proBNP (пмоль/л), среднее значение (СО) | 82.0 (159.2) | 54.0 (122.6) |
| Краткий опросник состояния здоровья, состоящий из 36 пунктов (SF-36), суммарный балл физического компонента, среднее значение (СО) | 37.2 (9.8) | 39.7 (9.3) |
| Суммарный балл симптомов и изменений нейропатии (NSC), среднее значение (СО) | 23.0 (12.6) | 23.1 (12.4) |
| Показатель инвалидизации при полинейропатии (PND), n (%) | ||
| I | 23 (38.3) | 56 (39.2) |
| II | 19 (31.7) | 61 (42.7) |
| IIIa | 15 (25.0) | 16 (11.2) |
| IIIb | 3 (5.0) | 10 (7.0) |
| Индекс массы тела (кг/м2) | ||
| m | 60 | 138 |
| Среднее значение (СО) | 24.2 (4.9) | 24.4 (4.9) |
| Медиана (мин., макс.) | 23.8 (14.5, 39.8) | 24.1 (15.4, 35.4) |
| Модифицированный индекс массы тела (кг/м2×г/л) | ||
| m | 60 | 138 |
| Среднее значение (СО) | 1049.89 (228.43) | 1025.78 (235.12) |
| Медиана (мин., макс.) | 1027.55 (668.7, 1710.0) | 1003.14 (615.7, 1714.0) |
* Группа с внешним плацебо из другого рандомизированного контролируемого исследования (NEURO-TTR).
1 Стадия заболевания определяется как стадия 1 = не требует помощи при передвижении и стадия 2 = требует помощи при передвижении.
2 Включает генотипы V30M, V50M, V50M MUTATION, VAL50MET и P.VAL50MET.
3 На основе клинической базы данных. Мутации, не относящиеся к Val30Met, включают: GLU89GLN, LEU58HIS, PHE64LEU, SER50ARG, SER77TYR, THR49ALA, THR60ALA, VAL122LLE и другие (включая ALA97SER).
4 Семейная амилоидная кардиомиопатия - наследственный транстиретиновый амилоидоз с кардиомиопатией (ATTRv-CM).
5 Знаменателем при расчете процента является количество пациентов с диагнозом FAC.
Для расчета продолжительности заболевания с момента постановки диагноза и от появления симптомов ATTRv-PN, FAC собирали данные только по году и месяцам, начиная с даты подписания информированного согласия.
N - количество пациентов в популяции для оценки безопасности; n - количество пациентов в подгруппе, m - количество пациентов с отсутствующими данными, если они отличаются от N, ИРК - индивидуальная регистрационная карта; NT-proBNP - N-терминальный пропептид натрийуретического гормона В-типа; СО – стандартное отклонение.
Анализ на 35-й неделе (промежуточный анализ)
Первичными конечными точками эффективности являлись изменение концентрации TTR в сыворотке крови относительно исходного уровня до 35-й недели (см. рисунок 2) и комбинированной оценки по модифицированной шкале нарушений нейропатии +7 (mNIS+7). Комбинированная оценка по mNIS+7 является объективной оценкой нейропатии и включает в себя комбинированную оценку по NIS и Modified +7. В версии комбинированной оценки по mNIS+7, использованной в исследовании, по шкале NIS объективно измеряют нарушение функции черепно-мозговых нервов, мышечной силы, рефлексов и ощущений, а по Modified +7 оценивают ответ со стороны сердечных сокращений на глубокое дыхание, количественное сенсорное тестирование (прикосновение-давление и тепло-боль), а также электрофизиологию периферических нервов. Аналитическая область валидированной версии комбинированной оценки по mNIS+7, использованной в исследовании, составляла от -22.3 до 346.3 баллов, причем более высокие баллы отражали большую степень тяжести заболевания.
Вторичной конечной точкой являлось изменение общего балла по Норфолкскому опроснику качества жизни - диабетическая нейропатия (QoL-DN) по сравнению с исходным уровнем. Шкала Норфолк QoL-DN представляет собой оценку пациентом субъективных ощущений от нейропатии в следующих областях: физическое функционирование/нейропатия крупных волокон, повседневная деятельность, симптомы, нейропатия мелких волокон и вегетативная нейропатия. Версия общей оценки по шкале Норфолк QoL-DN, использовавшаяся в исследовании, имела диапазон от -4 до 136 баллов, причем более высокие баллы отражали более выраженные нарушения.
При применении препарата Ирелми установлено статистически значимое улучшение по сравнению с внешним плацебо на 35-й неделе в отношении снижения уровня TTR в сыворотке крови с процентным изменением -66.43% (95% ДИ: -71.39%, -61.47%; p< 0.0001) (см. рисунок 2). При применении препарата Ирелми установлено статистически значимое улучшение по сравнению с внешним плацебо на 35-й неделе по комбинированному баллу mNIS+7 с различием в среднем НК -9.0 (95% ДИ: -13.5, -4.5; p< 0.0001) (см. рисунки 3, 4а, 7а). При применении препарата Ирелми установлено статистически значимое улучшение по сравнению с внешним плацебо на 35-й неделе по суммарному баллу по шкале Норфолк QoL-DN с различием средних значений, полученных методом НК, -11.8 (95% ДИ: -16.8, -6.8; p< 0.0001) (таблица 2 и рисунки 5, 6a, 8a).
Анализ на 65/66-й неделе (итоговый анализ)
К комбинированным первичным конечным точкам для достижения основной цели при итоговом анализе на 66-й неделе относились процентное изменение по сравнению с исходным уровнем концентрации TTR в сыворотке крови на 65-й неделе, изменение по сравнению с исходным уровнем комбинированного балла по mNIS+7 на 66-й неделе и изменение по сравнению с исходным уровнем суммарного балла по Норфолк QoL-DN на 66-й неделе. На 65-й неделе снижение концентрации TTR в сыворотке крови сохранялось. Кроме того, результаты на 66-й неделе по комбинированному баллу по mNIS+7 и суммарному баллу по Норфолк соответствовали результатам на 35-й неделе (см. таблицу 2 и рисунки 3, 4б, 5, 6б).
Вторичными конечными точками являлись изменение по сравнению с исходным уровнем симптомов и изменений нейропатии (NSC) на 66-й и 35-й неделях, изменение по сравнению с исходным уровнем показателя физического компонента (PCS) краткого опросника состояния здоровья, состоящего из 36 пунктов (версия 2) (SF-36), на 65-й неделе, изменение по сравнению с исходным уровнем показателя инвалидизации при полинейропатии (PND) на 65-й неделе и изменение по сравнению с исходным уровнем модифицированного индекса массы тела (мИМТ) на 65-й неделе.
NSC представляет собой опросник, заполняемый пациентом и предназначенный для количественной оценки типа, распределения и тяжести мышечной слабости, сенсорных симптомов, болевых симптомов и вегетативных симптомов. Более высокие баллы означают более выраженные симптомы.
Опросник SF-36 PCS включает 4 шкалы, по которым оценивают физическую функцию, ролевое функционирование, обусловленное нарушением физического состояния, боль в теле и общее состояние здоровья. Более высокие баллы свидетельствуют о лучшем физическом здоровье.
В PND инвалидизация классифицирована по степени мобильности (например, необходимость использования трости, костыля, инвалидного кресла или кровати). Более высокий балл по шкале PND означает более тяжелую инвалидизацию.
мИМТ (ИМТ×концентрация альбумина в сыворотке крови) является приемлемым методом оценки состояния питания при ATTR. Более высокие баллы отражают лучшее состояние питания и считаются показателем более длительной выживаемости у пациентов с ATTRv-PN.
По всем вторичным конечным точкам было установлено статистически значимое превосходство над внешним плацебо (см. таблицу 3).
Таблица 2. Эффект лечения для первичной и ключевых вторичных конечных точек (исследование NEURO-TTRansform) (популяция полного анализа)
| Анализ/конечная точка | Исходное среднее значение (СО) | Изменение среднего НК/процентное изменение относительно исходного значения, (СОШ) [95% ДИ] | Ирелми - внешнее плацебо* Различие средних НК (95% ДИ) | Значение Р | ||
| Внешнее плацебо* | Ирелми | Внешнее плацебо* | Ирелми | |||
| Неделя 35 | N=59 | N=140 | N=59 | N=140 | ||
| Концентрация TTR в сыворотке крови, г/л1 | 0.15 (0.04) | 0.23 (0.08) | ||||
| Изменение (%) относительно исходного значения | -14.8% (2.0) [-18.73, -10.80] | -81.2% (1.7) [-84.55, -77.84] | -66.4% (71.39, -61.47) | Р<0.0001 | ||
| Комбинированный балл по mNIS+72,3 | 74.1 (39.0) | 79.6 (42.3) | ||||
| Изменение относительно исходного значения | 9.2 (1.9) [5.54, 12.91] | 0.2 (1.9) [-3.46, 3.89] | -9.0 (-13.48, -4.54) | Р<0.0001 | ||
| Суммарный балл по Норфолк QoL-DN2,3 | 48.6 (27.0) | 43.5 (26.3) | ||||
| Изменение относительно исходного значения | 8.7 (2.1) [4.53, 12.81] | -3.1 (2.1) [-7.19, 0.96] | -11.8 (-16.82, -6.76) | Р<0.0001 | ||
| Неделя 65/66 | N=59 | N=141 | N=59 | N=141 | ||
| Концентрация TTR в сыворотке крови, г/л1 | 0.15 (0.04) | 0.23 (0.08) | ||||
| Изменение (%) относительно исходного значения | -11.2% (1.9) [-15.06, -7.41] | -81.7% (1.6) [-84.82, -78.48] | -70.4% (-75.17, -65.66) | Р<0.00014 | ||
| Комбинированный балл по mNIS+71 | 74.1 (39.0) | 79.8 (42.3) | ||||
| Изменение относительно исходного значения | 25.1 (2.4) [20.23, 29.88] | 0.3 (2.4) [-4.46, 5.06] | -24.8 (-30.96, -18.56) | Р<0.00014 | ||
| Суммарный балл по Норфолк QoL-DN1 | 48.6 (27.0) | 43.3 (26.2) | ||||
| Изменение относительно исходного значения | 14.2 (2.4) [9.51, 18.97] | -5.5 (2.3) [-10.03, -0,96] | -19.7 (-25.63, -13.84) | Р<0.0001 | ||
* Группа с внешним плацебо из другого рандомизированного контролируемого исследования (NEURO-TTR).
1 На основе MMRM, скорректированной с помощью показателей предрасположенности с фиксированными категориальными эффектами для лечения, времени, взаимосвязи между лечением и временем, а также стадии заболевания, мутации Val30M, предшествующего лечения и фиксированных ковариат для исходного значения и взаимосвязи между исходным уровнем и временем. В промежуточный анализ на 66-й неделе включены только данные до 66-й недели.
2 На основе модели ковариационного анализа (ANCOVA), скорректированной с помощью показателей предрасположенности с эффектами лечения, стадии заболевания, мутации Val30M, предшествующего лечения и исходного значения. В промежуточный анализ включены только данные до 35-й недели.
3 Участникам с отсутствующими данными по mNIS+7 или Норфолк QoL-DN на 35-й неделе исследования значения были многократно подставлены с помощью модели восстановления пропущенных данных. Каждый из 500 подставленных наборов данных был проанализирован с помощью простой модели ковариационного анализа, а результаты 500 моделей ковариационного анализа были объединены с помощью правил Рубина.
4 Формально не определяли в связи с получением статистически значимых результатов на 35-й неделе.
Анализ проводили на основании данных, собранных в течение периода до 52 дней после применения последней дозы исследуемого препарата. Данные 35-й недели - из промежуточного анализа, а данные 65/66-й недели - из анализа на 66-й неделе. В популяции полного анализа группа эплонтерсена включала 140 участников на 35-й неделе и 141 участника на 66-й неделе. У одного участника отсутствовала оценка по mNIS+7 или Норфолк QoL-DN на 35-й неделе, но была оценка хотя бы по одному из этих показателей на 66-й неделе.
ANCOVA - ковариационный анализ; ДИ – доверительный интервал; среднее НК – среднее значение, рассчитанное по методу наименьших квадратов; MMRM – модель смешанных эффектов с повторными измерениями; mNIS+7 – модифицированная шкала нарушений нейропатии +7 баллов; N – количество участников в группе; Норфолк QoL-DN – Норфолкский опросник качества жизни – диабетическая нейропатия; СО – стандартное отклонение; СОШ – стандартная ошибка; TTR – транстиретин.
Таблица 3. Иерархическое тестирование вторичных конечных точек (исследование NEURO-TTRansform)
| Вторичная конечная точка/группа лечения (N) | n | Изменение среднего НК относительно исходного уровня (95% ДИ) | Сравнение препарата Ирелми с внешним плацебо* | ||
| Рассчитанное значение | 95% ДИ | Значение p | |||
| Изменение среднего НК в NSC по сравнению с исходным уровнем на 66-й неделе | |||||
| Ирелми (N=141) | 132 | 0.0 (-1.92, 1.86) | -8.2 | -10.65, -5.76 | <0.0001 |
| Внешнее плацебо* (N=59) | 52 | 8.2 (6.24, 10.12) | |||
| Изменение среднего НК в NSC по сравнению с исходным уровнем на 35-й неделе | |||||
| Ирелми (N=141) | 141 | 0.8 (-0.92, 2.50) | -3.9 | -6.08, -1.80 | 0.0005 |
| Внешнее плацебо* (N=59) | 56 | 4.7 (2.98, 6.48) | |||
| Изменение среднего НК в SF-36 PCS по сравнению с исходным уровнем на 65-й неделе | |||||
| Ирелми (N=141) | 136 | 0.85 (-0.711, 2.412) | 5.31 | 3.195, 7.416 | <0.0001 |
| Внешнее плацебо* (N=59) | 50 | -4.46 (-6.139, -2.770) | |||
| Изменение среднего НК в оценке по PND по сравнению с исходным уровнем на 65-й неделе | |||||
| Ирелми (N=141) | 134 | 0.1 (0.0, 0.2) | -0.2 | -0.4, 0.0 | 0.0241 |
| Внешнее плацебо* (N=59) | 51 | 0.3 (0.2, 0.4) | |||
| Изменение среднего НК в мИМТ по сравнению с исходным уровнем на 65-й неделе | |||||
| Ирелми (N=141) | 130 | -8.1 (-28.55, 12.42) | 82.7 | 54.64, 110.76 | <0.0001 |
| Внешнее плацебо* (N=59) | 49 | -90.8 (-112.84, -68.69) | |||
* Группа с внешним плацебо из другого рандомизированного контролируемого исследования (NEURO-TTR).
N – количество пациентов в популяции полного анализа на 66-й неделе.
n – количество пациентов с отсутствующими данными по исходным ковариатам и изменению по сравнению с исходным уровнем в данной временной точке.
Анализ проводили на основании данных, собранных в течение периода до 28 дней после применения последней дозы исследуемого препарата. Временные рамки визитов в рамках анализа на 65-й неделе – с 419 по 479-й день.
На основе модели со смешанными эффектами для повторных измерений (MMRM), скорректированной с помощью показателей предрасположенности с фиксированными категориальными эффектами для лечения, времени, взаимосвязи между лечением и временем, а также стадии заболевания, мутации Val30M, предшествующего лечения и фиксированных ковариат для исходного значения и взаимосвязи между исходным уровнем и временем. В итоговый анализ на 66-й неделе включены только данные до 65-й недели.
ДИ – доверительный интервал; среднее НК – среднее значение, рассчитанное по методу наименьших квадратов; мИМТ – модифицированный индекс массы тела; NSC – симптомы и изменения нейропатии; PND – инвалидизация при полинейропатии; PCS – оценка физического компонента; SF-36 PCS = краткий опросник оценки состояния здоровья по физическому компоненту, состоящий из 36 пунктов.
Рисунок 2. Процентное изменение концентрации TTR в сыворотке крови относительно исходного уровня до 65-й недели, препарат Ирелми по сравнению с внешним плацебо* на 65-й неделе (исследование NEURO-TTRansform) (популяция полного анализа)
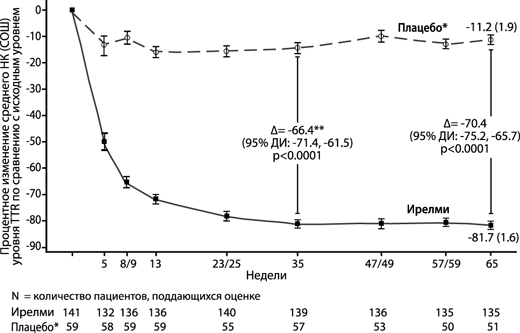
* Группа с внешним плацебо из другого рандомизированного контролируемого исследования (NEURO-TTR).
** Различие между видами лечения представляет результаты формального промежуточного анализа на 35-й неделе. В промежуточный анализ на 35-й неделе включены только данные до 35-й недели.
На основе MMRM, скорректированной с помощью показателей предрасположенности с фиксированными категориальными эффектами для лечения, времени, взаимосвязи между лечением и временем, а также стадии заболевания, мутации Val30Met, предшествующего лечения и фиксированных ковариаций для исходного уровня и взаимосвязи между исходным уровнем и временем.
Анализ проводили на основании данных, собранных в течение периода до 28 дней после применения последней дозы исследуемого препарата. Включены данные до 65-й недели. Плацебо оценивали на исходном уровне, на неделях 5, 8, 13, 23, 35, 47, 57 и 65, препарат Ирелми – на исходном уровне, на неделях 5, 9, 13, 25, 35, 49, 59 и 65.
Представлено различие между группами лечения в отношении средних НК на 35-й неделе и 65-й неделе (препарат Ирелми – плацебо) с 95% ДИ (нескорректированное).
ДИ – доверительный интервал; среднее НК – среднее значение, рассчитанное по методу наименьших квадратов; СОШ – стандартная ошибка среднего, MMRM – модель смешанных эффектов с повторными измерениями; TTR – транстиретин.
Рисунок 3. Изменение среднего НК в комбинированной оценке по mNIS+7 по сравнению с исходным уровнем (исследование NEURO-TTRansform) (популяция полного анализа)
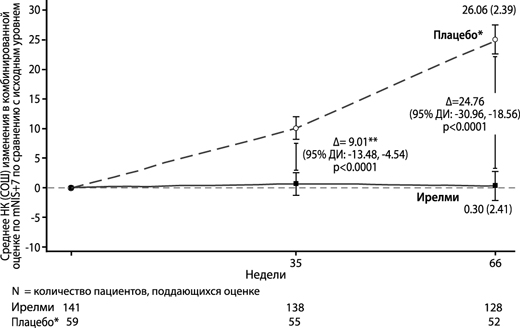
* Группа с внешним плацебо из другого рандомизированного контролируемого исследования (NEURO-TTR).
** Различие между видами лечения представляет результаты формального промежуточного анализа на 35-й неделе. На основе MI ANCOVA, скорректированного с помощью показателей предрасположенности с фиксированными категориальными эффектами для лечения, стадии заболевания, мутации Val30Met, предшествующего лечения и фиксированными ковариатами для исходного уровня. В промежуточный анализ на 35-й неделе включены только данные до 35-й недели.
Анализ на 66-й неделе на основе MMRM, скорректированной с помощью показателей предрасположенности с категориальными эффектами для лечения, времени, взаимосвязи между лечением и временем, а также стадии заболевания, мутации Val30Met, предшествующего лечения и фиксированных ковариат для исходного уровня и взаимосвязи между исходным уровнем и временем.
Анализ проводили на основании данных, собранных в течение периода до 52 дней после применения последней дозы исследуемого препарата. Включены данные до 66-й недели.
Представлено различие между группами лечения в отношении средних НК на 35-й неделе и 65-й неделе (препарат Ирелми – плацебо) с 95% ДИ (нескорректированное).
ДИ – доверительный интервал; среднее НК – среднее значение, рассчитанное по методу наименьших квадратов; СОШ – стандартная ошибка среднего, MI ANCOVA = ковариационный анализ по методу множественной подстановки; MMRM – модель смешанных эффектов с повторными измерениями.
Рисунок 4. Гистограмма изменения комбинированного балла по mNIS+7 по сравнению с исходным уровнем (исследование NEURO-TTRansform) (популяция полного анализа)
а) на 35-й неделе
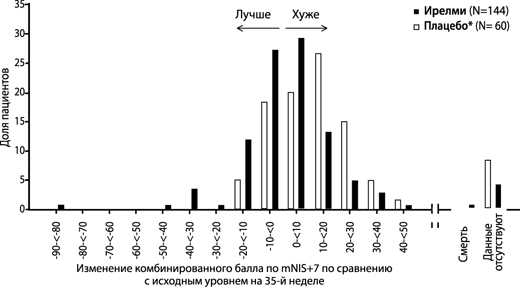
* Группа с внешним плацебо из другого рандомизированного контролируемого исследования (NEURO-TTR).
б) на 66-й неделе

* Группа с внешним плацебо из другого рандомизированного контролируемого исследования (NEURO-TTR).
Рисунок 5. Изменение среднего НК общего балла по шкале Норфолк QoL-DN по сравнению с исходным уровнем (исследование NEURO-TTRansform)
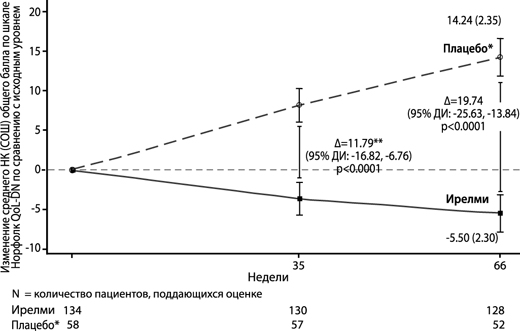
* Группа с внешним плацебо из другого рандомизированного контролируемого исследования (NEURO-TTR).
** Различие между видами лечения представляет результаты формального промежуточного анализа на 35-й неделе. На основе MI ANCOVA, скорректированного с помощью показателей предрасположенности с фиксированными категориальными эффектами для лечения, стадии заболевания, мутации Val30Met, предшествующего лечения и фиксированными ковариатами для исходного уровня. В промежуточный анализ на 35-й неделе включены только данные до 35-й недели.
Анализ на 66-й неделе на основе MMRM, скорректированной с помощью показателей предрасположенности с категориальными эффектами для лечения, времени, взаимосвязи между лечением и временем, а также стадии заболевания, мутации Val30Met, предшествующего лечения и фиксированных ковариат для исходного уровня и взаимосвязи между исходным уровнем и временем.
Анализ проводили на основании данных, собранных в течение периода до 52 дней после применения последней дозы исследуемого препарата. Включены данные до 66-й недели.
Представлено различие между группами лечения в отношении средних НК на 35-й неделе и 65-й неделе (Ирелми – плацебо) с 95% ДИ (нескорректированное).
ДИ – доверительный интервал; среднее НК – среднее значение, рассчитанное по методу наименьших квадратов; СОШ – стандартная ошибка среднего, MI ANCOVA – ковариационный анализ по методу множественной подстановки; MMRM – модель смешанных эффектов с повторными измерениями.
Рисунок 6. Гистограмма изменения общего балла по шкале Норфолк QoL-DN по сравнению с исходным уровнем (исследование NEURO-TTRansform) (популяция полного анализа)
а) на 35-й неделе

* Группа с внешним плацебо из другого рандомизированного контролируемого исследования (NEURO-TTR).
б) на 66-й неделе
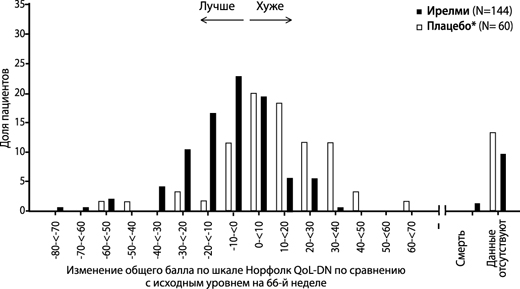
* Группа с внешним плацебо из другого рандомизированного контролируемого исследования (NEURO-TTR).
На 35-й и 65/66-й неделях у пациентов, получавших препарат Ирелми, было отмечено сходное снижение концентрации TTR в сыворотке крови, улучшение комбинированного балла по mNIS+7 и общего балла по Норфолк QoL-DN по сравнению с плацебо во всех подгруппах, включая возраст, пол, расовую принадлежность, регион, исходный балл по NIS на исходном уровне, статус мутации Val30Met, статус кардиомиопатии, исходный диагноз FAC и стадию заболевания (рисунки 1a и б, 7a и б, 8a и б).
Рисунок 7. Форест-диаграмма различия между группами лечения в среднем НК для изменения комбинированного балла по mNIS+7 относительно исходного уровня в основных подгруппах (исследование NEURO-TTRansform) (популяция полного анализа)
а) на 35-й неделе

* Группа с внешним плацебо из другого рандомизированного контролируемого исследования (NEURO-TTR).
[a] Ранее получавшие лечение тафамидисом или дифлунисалом.
В подгруппу КМ вошли пациенты с диагнозом FAC на момент включения в исследование или исходной толщиной межжелудочковой перегородки ≥13 мм при отсутствии артериальной гипертензии (в анамнезе или диагностированной во время исследования).
Различие средних НК, доверительные интервалы и значения p основаны на модели ANCOVA, скорректированной с помощью показателей предрасположенности с эффектами лечения, факторов подгрупп, стадии заболевания, мутации Val30Met, предшествующего лечения, взаимосвязи между лечением и подгруппой, а также исходного значения. В промежуточный анализ на 35-й неделе включены данные до 35-й недели.
б) на 66-й неделе
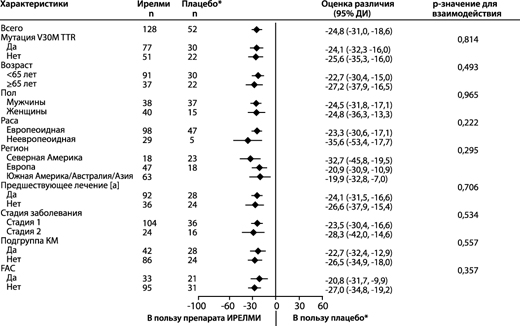
* Группа с внешним плацебо из другого рандомизированного контролируемого исследования (NEURO-TTR).
[a] Ранее получавшие лечение тафамидисом или дифлунисалом.
На основе MMRM, скорректированной с помощью показателей предрасположенности с категориальными эффектами для лечения, времени, взаимосвязи между лечением и временем, а также стадии заболевания, мутации Val30Met, предшествующего лечения и фиксированных ковариат для исходного уровня и взаимосвязи между исходным уровнем и временем.
Модели подгрупп также включали взаимосвязи между лечением и подгруппой, временем и подгруппой и лечением, временем и подгруппой. Включены данные до 66-й недели.
В подгруппу КМ вошли пациенты с диагнозом FAC на момент включения в исследование или исходной толщиной межжелудочковой перегородки ≥13 мм при отсутствии артериальной гипертензии [в анамнезе или диагностированной во время исследования].
Представлено различие между группами лечения средних НК на 66-й неделе (препарат Ирелми – плацебо) с 95% ДИ (нескорректированное). ДИ – доверительный интервал; среднее НК – среднее значение, рассчитанное по методу наименьших квадратов; MMRM – модель смешанных эффектов с повторными измерениями; КМ – кардиомиопатия, FAC – семейная амилоидная кардиомиопатия.
Рисунок 8. Форест-диаграмма различия между группами лечения в отношении средних НК для изменения общего балла по Норфолк QoL-DN относительно исходного уровня в основных подгруппах (исследование NEURO-TTRansform) (популяция полного анализа)
а) на 35-й неделе
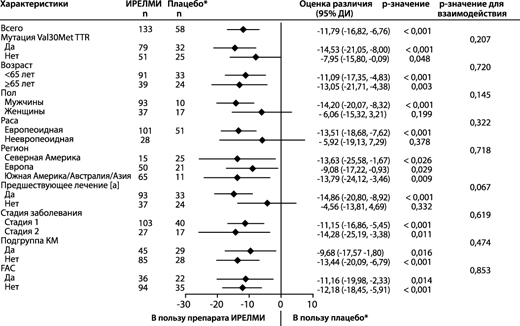
* Группа с внешним плацебо из другого рандомизированного контролируемого исследования (NEURO-TTR).
[a] Ранее получавшие лечение тафамидисом или дифлунисалом.
В подгруппу КМ вошли пациенты с диагнозом FAC на момент включения в исследование или исходной толщиной межжелудочковой перегородки ≥13 мм при отсутствии артериальной гипертензии (в анамнезе или диагностированной во время исследования).
Различие средних НК, доверительные интервалы и p-значения основаны на модели ANCOVA, скорректированной с помощью показателей предрасположенности с эффектами лечения, факторов подгрупп, стадии заболевания, мутации Val30Met, предшествующего лечения, взаимосвязи между лечением и подгруппой, а также исходного значения. В промежуточный анализ на 35-й неделе включены только данные до 35-й недели.
б) на 66-й неделе
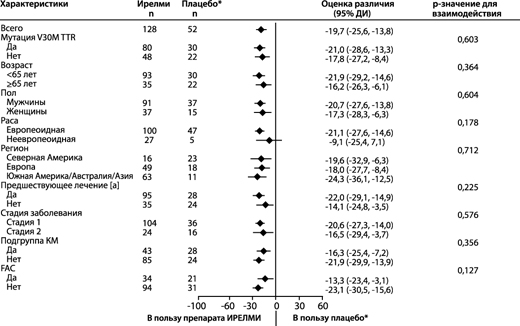
* Группа с внешним плацебо из другого рандомизированного контролируемого исследования (NEURO-TTR).
[a] Ранее получавшие лечение тафамидисом или дифлунисалом.
На основе MMRM, скорректированной с помощью показателей предрасположенности с категориальными эффектами для лечения, времени, взаимосвязи между лечением и временем, а также стадии заболевания, мутации Val30Met, предшествующего лечения и фиксированных ковариат для исходного уровня и взаимосвязи между исходным уровнем и временем.
Модели подгрупп также включали взаимосвязи между лечением и подгруппой, временем и подгруппой и лечением, временем и подгруппой. Включены данные до 66-й недели.
В подгруппу КМ вошли пациенты с диагнозом FAC на момент включения в исследование или исходной толщиной межжелудочковой перегородки ≥13 мм при отсутствии артериальной гипертензии [в анамнезе или диагностированной во время исследования].
Представлено различие между группами лечения средних НК на 66-й неделе (Ирелми – плацебо) с 95% ДИ (нескорректированное). ДИ – доверительный интервал; среднее НК – среднее значение, рассчитанное по методу наименьших квадратов; MMRM – модель смешанных эффектов с повторными измерениями; КМ – кардиомиопатия, FAC – семейная амилоидная кардиомиопатия.
В ходе поискового анализа оценки состояния сердца с помощью серийных эхокардиограмм при применении препарата Ирелми установлено улучшение соотношения E/e' (показатель диастолической функции левого желудочка) после 65 недель лечения в подгруппе кардиомиопатии (скорректированное плацебо-контролируемое различие средних НК: -3.94 [95% ДИ -6.46, -1.42]). Направленные изменения в сторону преимущества препарата Ирелми по сравнению с плацебо на 66-й неделе также были отмечены по предварительно определенным поисковым кардиологическим конечным точкам в виде средней толщины стенки левого желудочка (различие в среднем НК -0.04 см, [95% ДИ -0.12, 0.04]), толщины стенок межжелудочковой перегородки (различие -0.05 см, [95% ДИ -0.16, 0.06]) и NT-proBNP, прогностического биомаркера нарушения функции сердца (геометрическое среднее НК 0.88, [95% ДИ 0.68, 1.14]). Несмотря на эти полученные значения, клиническая польза при кардиомиопатии пока не подтверждена.
Анализ на 85-й неделе (анализ по окончании лечения)
Данные по 85-й неделе для группы внешнего плацебо отсутствуют, поскольку период лечения в исследовании NEURO-TTR составил всего 66 недель.
Наблюдаемый эффект в группе лечения препаратом Ирелми в отношении комбинированного балла по mNIS+7 являлся устойчивым и сохранялся до конца лечения на 85-й неделе. Среднее (СО) изменение комбинированного балла по mNIS+7 по сравнению с исходным уровнем составило -0.04% (16.2) на 35-й неделе, -0.21% (17.6) на 66-й неделе и -2.9% (20.5) на 85-й неделе. Средний общий балл по шкале Норфолк QoL-DN оставался стабильным до 85-й недели. В группе эплонтерсена среднее (СО) изменение относительно исходного уровня общего балла по шкале Норфолк QoL-DN составило -4.8 (16.5) на 35-й неделе, -7.2 (18.5) на 66-й неделе и -6.2 (18.0) на 85-й неделе.
Результаты оценки по NSC, PND и мИМТ оставались стабильными до 85-й недели, а по шкале SF-36 сохранялась тенденция к улучшению.
Данные доклинической безопасности
Токсичность при многократном введении в доклинических исследованиях
Многократное применение эплонтерсена или его суррогата для грызунов приводило к снижению уровня мРНК TTR в печени (до ≈62% и 82% у обезьян и мышей соответственно) с последующим снижением уровня белка TTR в плазме крови (до 70% у обезьян). Токсикологически значимых результатов, связанных с фармакологическим ингибированием экспрессии TTR, не обнаружено.
Большинство изменений, наблюдавшихся после многократного применения препарата в течение 6 месяцев у мышей и 9 месяцев у обезьян, были связаны с поглощением и накоплением эплонтерсена и не считались нежелательными. Микроскопические изменения, связанные с поглощением эплонтерсена, были отмечены у различных типов клеток в различных органах всех исследованных видов животных, включая моноциты/макрофаги, эпителий проксимальных канальцев почек, клетки Купфера в печени и инфильтраты гистиоцитарных клеток в лимфатических узлах и местах инъекций.
В исследовании субхронической токсичности у одной обезьяны в максимальной дозе (24 мг/кг/неделя) было отмечено резкое снижение числа тромбоцитов, сопровождавшееся спонтанными кровоизлияниями. Сходные изменения были отмечены у обезьян, получавших препарат в средней дозе 6 мг/кг/неделя, что в 73 раза превышает AUC человека в рекомендуемой терапевтической дозе эплонтерсена.
Мутагенность и канцерогенность
Эплонтерсен не проявлял генотоксического потенциала in vitro и in vivo и не являлся канцерогенным для трансгенных мышей ras.H2.
Эплонтерсен не продемонстрировал генотоксичности в анализах in vitro (бактериальная мутагенность, хромосомные аберрации в легких китайского хомячка) и in vivo (микроядра костного мозга мыши).
В исследовании канцерогенности при подкожном введении на трансгенных мышах ras.H2 эплонтерсен вводили в течение 26 недель в дозах 250, 500 и 1500 мг/кг/месяц. После лечения мышей в течение 26 недель признаков канцерогенности эплонтерсена не обнаружено.
Репродуктивная токсичность
Эмбриофетальная/онтогенетическая токсичность/фертильность
Применение эплонтерсена не оказывало влияния на фертильность и развитие эмбриона и плода у мышей в дозе, до 38 раз превышающей рекомендуемую дозу 45 мг в месяц для человека. Эплонтерсен не проявляет фармакологической активности у мышей. В то же время, при применении у мышей аналога эплонтерсена, который вызывал ингибирование экспрессии мРНК TTR на >90%, влияния на фертильность и развитие эмбриона и плода тоже не наблюдалось.
Фармакокинетика
Фармакокинетические (ФК) свойства препарата Ирелми оценивали после п/к введения однократных и многократных доз (1 раз в 4 недели) здоровым добровольцам и многократных доз (1 раз в 4 недели) пациентам с ATTRv-PN.
Всасывание
После п/к введения эплонтерсен быстро всасывается в системный кровоток, время достижения Cmax в плазме крови составляет приблизительно 2 ч согласно популяционным оценкам.
Распределение
По данным исследований на животных (мыши, крысы и обезьяны), после п/к введения эплонтерсен распределяется преимущественно в корковом веществе печени и почек. Эплонтерсен обладает высокой степенью связывания с белками плазмы крови человека (>98%). Популяционные оценки для кажущегося Vd в центральном компартменте составляют 12.9 л, а кажущегося Vd в периферическом компартменте - 11100 л.
Метаболизм
Эплонтерсен метаболизируется эндо- и экзонуклеазами до коротких олигонуклеотидных фрагментов разного размера в печени. Крупные (длинноцепочечные) циркулирующие метаболиты в организме человека не обнаружены. Олигонуклеотидные препараты, включая эплонтерсен, как правило, не метаболизируются ферментами цитохрома P (CYP).
Выведение
Эплонтерсен выводится преимущественно путем метаболизма с последующей почечной экскрецией коротких олигонуклеотидных метаболитов. Средняя доля неизмененного ASO, выведенного с мочой, составила менее 1% от введенной дозы в течение 24 ч. Согласно популяционным оценкам, конечный T1/2 составляет приблизительно 3 недели.
Линейность/нелинейность
Равновесная Cmax и AUC эплонтерсена несколько превышали дозопропорциональное увеличение после однократного п/к введения препарата в дозах от 45 до 120 мг (т.е. в 1–2.7 раза выше рекомендуемой дозы) у здоровых добровольцев.
Популяционные оценки Cmax, минимальных концентраций (Ctrough) и AUC составили 0.218 мкг/мл, 0.000200 мкг/мл и 1.95 мкг×ч/мл соответственно после применения препарата в дозе 45 мг 1 раз в 4 недели у пациентов с ATTRv-PN. При многократном введении препарата (1 раз в 4 недели) накопления эплонтерсена в плазме крови при Cmax и AUC не наблюдали. Накопление было отмечено для Ctrough, а равновесная концентрация достигается примерно через 17 недель.
Фармакокинетика у особых групп пациентов
Пол, раса, масса тела
По данным популяционного фармакокинетического и фармакодинамического анализов, масса тела, пол, расовая принадлежность и статус мутации Val30Met не оказывают клинически значимого влияния на экспозицию эплонтерсена и снижение уровня TTR в сыворотке крови в равновесном состоянии. В некоторых случаях окончательные оценки были ограничены, т.к. ковариаты были ограничены общим низким количеством.
Лица пожилого возраста
В целом, различий в фармакокинетике между взрослыми пациентами и лицами пожилого возраста (≥65 лет) не отмечено.
Нарушение функции почек
Формальных клинических исследований по изучению влияния почечной недостаточности на ФК эплонтерсена не проводили. При проведении популяционного фармакокинетического и фармакодинамического анализов не выявлено клинически значимых различий в фармакокинетике и фармакодинамике эплонтерсена при почечной недостаточности легкой и умеренной степени (рСКФ от ≥30 до <90 мл/мин.). Применение эплонтерсена не изучали у пациентов с тяжелой почечной недостаточностью или у пациентов с терминальной стадией почечной недостаточности.
Нарушение функции печени
Формальные клинические исследования по изучению влияния печеночной недостаточности на эплонтерсен не проводили. При проведении популяционного фармакокинетического и фармакодинамического анализов не выявлено клинически значимых различий в фармакокинетике и фармакодинамике эплонтерсена при печеночной недостаточности легкой степени (общий билирубин ≤1×ВГН и АСТ >1×ВГН или общий билирубин >1.0-1.5×ВГН и любая АСТ). Применение эплонтерсена не изучали у пациентов с печеночной недостаточностью умеренной или тяжелой степени, а также у пациентов с предшествующей трансплантацией печени.
Лекарственные взаимодействия
Исследований взаимодействия с лекарственными препаратами не проводили. В исследованиях in vitro установлено, что эплонтерсен не является субстратом или ингибитором транспортеров, не взаимодействует с препаратами, которые активно связываются с белками плазмы крови, не является ингибитором или индуктором ферментов CYP. Олигонуклеотидные препараты, включая эплонтерсен, как правило, не являются субстратами ферментов CYP. Таким образом, не ожидается, что применение эплонтерсена будет вызывать или влиять на взаимодействия с лекарственными препаратами, опосредованные через транспортеры препаратов, связывание с белками плазмы крови или ферментами CYP.
Иммуногенность
Наличие антител к эплонтерсену не влияло на Cmax и AUC эплонтерсена в плазме крови, но увеличивало Ctrough. У пациентов с наличием антител к эплонтерсену не было выявлено клинически значимого влияния на эффективность, безопасность, фармакокинетику и фармакодинамику препарата Ирелми.
Показания препарата Ирелми
- лечение взрослых пациентов в возрасте 18 лет и старше с полинейропатией, связанной с наследственным транстиретиновым амилоидозом (ATTRv).
Режим дозирования
Взрослые
Рекомендуемая доза препарата Ирелми составляет 45 мг. Препарат необходимо вводить в виде подкожной инъекции 1 раз в месяц.
Пропущенная доза
Если доза препарата пропущена, необходимо ввести ее как можно скорее. После этого следует возобновить применение препарата с интервалами в 1 месяц, начиная с даты введения последней дозы.
Особые группы пациентов
Пациенты пожилого возраста
Пожилым пациентам в возрасте 65 лет и старше коррекция дозы не требуется (см. раздел "Фармакокинетика").
Пациенты с нарушением функции почек
У пациентов с нарушением функции почек легкой и средней степени тяжести (рСКФ от ≥30 до <90 мл/мин/1.73 м2) коррекция дозы не требуется (см. раздел "Фармакокинетика"). Применение препарата Ирелми не изучали у пациентов с тяжелой почечной недостаточностью или с терминальной стадией почечной недостаточности.
Пациенты с нарушением функции печени
Пациентам с нарушением функции печени легкой степени тяжести коррекция дозы не требуется (общий билирубин ≤1×ВГН и АСТ >1×ВГН, или общий билирубин >1.0-1.5×ВГН и любой показатель АСТ) (см. раздел "Фармакокинетика"). Применение препарата Ирелми у пациентов с нарушением функции печени средней или тяжелой степени не изучали.
Дети
Безопасность и эффективность препарата Ирелми у детей и подростков в возрасте до 18 лет не установлены. Данные отсутствуют.
Способ применения
Препарат Ирелми вводится в виде подкожной инъекции.
Первую инъекцию, выполняемую пациентом или лицом, осуществляющим за ним уход, следует проводить под руководством медицинского работника с соответствующей квалификацией. Пациентов и/или лиц, осуществляющих за ними уход, следует обучить подкожному введению препарата Ирелми.
Шприц-ручку следует достать из холодильника не менее чем за 30 минут до введения препарата и дать ей нагреться до комнатной температуры перед инъекцией. Другие методы нагревания использовать не следует.
Перед применением следует визуально оценить раствор. Раствор должен быть прозрачным от бесцветного до желтого цвета. Не применять, если перед введением отмечаются помутнение, видимые частицы или изменение цвета раствора.
При самостоятельном применении препарат Ирелми вводят в область живота или верхней части бедра. При выполнении инъекции лицом, осуществляющим уход за пациентом, препарат также можно вводить в область задней поверхности плеча.
Инструкция по использованию препарата Ирелми
Пациенту следует прочитать данную инструкцию до начала использования шприц-ручки и всякий раз при применении препарата из новой пачки, т.к. информация может обновляться.
Важная информация
Шприц-ручку препарата Ирелми следует хранить в холодильнике при температуре от 2° до 8°C в оригинальной картонной упаковке до момента использования. При необходимости невскрытую упаковку препарата Ирелми можно хранить при комнатной температуре ниже 30°C не более 6 недель.
Каждая шприц-ручка содержит одну дозу и предназначена только для однократного применения.
Не следует использовать шприц-ручку, если:
- препарат был заморожен;
- шприц-ручку уронили, или она была повреждена, или контроль первого вскрытия на картонной пачке нарушен;
- срок годности препарата истек.
Не следует использовать шприц-ручку для других лиц.
Следует хранить шприц-ручку в недоступном для детей месте.
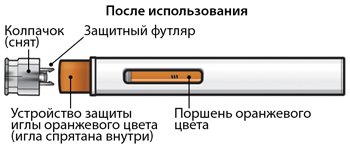
Инъекция начинается автоматически, когда устройство защиты иглы оранжевого цвета прижимают к коже.

Для получения полной дозы лекарственного препарата необходимо плотно прижать шприц-ручку к коже.
Инъекция считается выполненной только в том случае, если смотровое окно полностью оранжевое (не показано на рисунке).
Перед снятием защитного колпачка и введением препарата следует полностью прочитать инструкцию.
Шприц-ручка препарата Ирелми
Не следует снимать колпачок шприц-ручки до самого момента выполнения инъекции.
Не следует прикасаться к устройству защиты иглы оранжевого цвета.
Подготовка к инъекции препарата с помощью шприц-ручки
Шаг 1 - подготовить принадлежности для инъекции
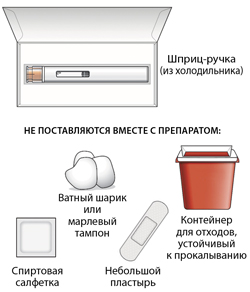
Шаг 2 - осмотреть упаковку и подождать 30 минут
До введения препарата необходимо выдержать картонную пачку с шприц-ручкой препарата в течение 30 минут при комнатной температуре.

Не следует подогревать шприц-ручку другими способами. Например, не следует разогревать ее в микроволновой печи или горячей воде, не следует класть рядом с источниками тепла.
Необходимо держать шприц-ручку вдали от света и прямых солнечных лучей.
Шаг 3 - извлечь шприц-ручку из упаковки и осмотреть ее
Проверить шприц-ручку на наличие повреждений.
Проверить дату истечения срока годности.
Осмотреть раствор через смотровое окно.
Допускается наличие в растворе небольших пузырьков воздуха.
Раствор должен быть прозрачным, от бесцветного до желтого цвета.
Не следует вводить препарат, если раствор мутный, имеет другой цвет или содержит видимые частицы.
Введение препарата с помощью шприц-ручки
Шаг 4 - выбрать место инъекции
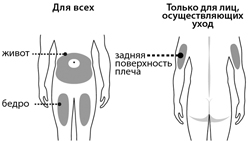
Рекомендуемое место введения препарата - передняя поверхность бедра или нижняя часть области живота (передняя брюшная стенка).
При выполнении инъекции лицом, осуществляющим уход за пациентом, препарат также можно вводить в область задней поверхности плеча. Если пациент вводит препарат самостоятельно, не следует вводить препарат в область плеча.
Для каждой последующей инъекции следует выбирать новое место на расстоянии не менее 3 см от места предшествующей инъекции.
Не следует вводить препарат:
- в радиусе 5 см вокруг пупка;
- в области, где кожа покраснела, имеет повышенную температуру; в участки чувствительной кожи; в область ушиба, шелушения или уплотнения кожи;
- в участки с рубцами, в область поврежденной, изменившей цвет или татуированной кожи;
- через одежду.
Шаг 5 - вымыть руки и обработать место инъекции
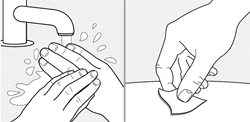
Тщательно вымыть руки водой с мылом.
Обработать место инъекции спиртовой салфеткой или водой с мылом. Дать ему высохнуть на воздухе.
Не следует касаться обработанной области до момента инъекции.
Шаг 6 - снять колпачок
Удерживая корпус шприц-ручки одной рукой, другой рукой аккуратно снять прозрачный колпачок с иглы. Теперь видно устройство защиты иглы оранжевого цвета, а игла скрыта за ним.
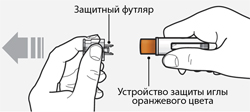
- Выбросить (утилизировать) прозрачный колпачок.
- Не следует прикасаться к игле, не следует нажимать пальцем на устройство защиты иглы оранжевого цвета.
- Не следует пытаться надеть колпачок обратно на шприц-ручку. Это может вызвать преждевременное высвобождение препарата или повреждение шприц-ручки.
Шаг 7 - введение препарата с помощью шприц-ручки
Ввести препарат с помощью шприц-ручки, следуя инструкциям на рисунках a, b, c и d. При введении препарата следует нажать и удерживать шприц-ручку в течение 10 секунд, пока оранжевый поршень не заполнит смотровое окно. Пациент может услышать первый "щелчок" в начале инъекции и второй "щелчок" в конце инъекции. Это нормально. Не следует перемещать и менять положение шприц-ручки после начала инъекции.
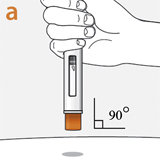
Положение шприц-ручки
Расположить устройство защиты иглы под прямым углом (90°) к поверхности кожи.
Пациент должен убедиться, что видит смотровое окно.
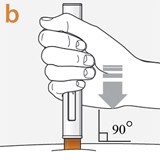
Плотно прижать шприц-ручку, пока устройство защиты иглы оранжевого цвета не скроется.
Будет слышен первый "щелчок". Щелчок означает, что введение препарата началось
В смотровом окне будет видно продвижение поршня оранжевого цвета вниз по мере введения препарата.
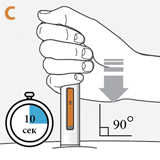
Плотно прижимая, удерживать в течение примерно 10 секунд.
Смотровое окно занимает поршень оранжевого цвета.
Будет слышен второй "щелчок". Второй щелчок означает, что введение препарата завершено.
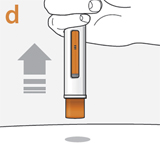
После завершения инъекции поднять шприц-ручку вертикально вверх
При этом устройство защиты иглы оранжевого цвета выйдет и займет место вокруг иглы (скроет иглу).
Шаг 8 - Проверить смотровое окно
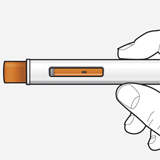
Проверить смотровое окно, чтобы убедиться, что раствор был введен полностью.
Если поршень оранжевого цвета не занимает смотровое окно полностью, возможно, пациент не получил полную дозу препарата. В таком случае, а также при возникновении других вопросов следует обратиться к лечащему врачу.
Шаг 9 - Проверить место инъекции
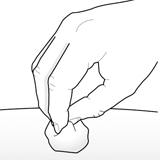
В месте инъекции может быть небольшое количество крови или жидкости. Это нормально.
При необходимости следует слегка прижать ватный шарик или марлевый тампон к месту инъекции и наложить небольшой пластырь.
Шаг 10 – утилизация использованной шприц-ручки
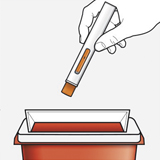
Сразу после использования следует поместить использованную шприц-ручку в контейнер для утилизации острых предметов, устойчивый к прокалыванию.
Не следует утилизировать шприц-ручку вместе с бытовыми отходами.
Рекомендации по утилизации
Необходимо утилизировать наполненный контейнер, следуя инструкциям работника аптеки.
Не следует направлять на переработку устойчивый к прокалыванию контейнер.
Побочное действие
Резюме профиля безопасности
Представленные ниже данные по безопасности были получены в клиническом исследовании при применении препарата Ирелми у 144 пациентов с полинейропатией, вызванной ATTRv (ATTRv-PN), рандомизированных для получения препарата Ирелми и получивших по меньшей мере одну дозу этого препарата. Из них 141 пациент получал лечение в течение не менее шести месяцев, а 137 пациентов - не менее 12 месяцев. Средняя продолжительность лечения составила 541 день (диапазон от 57 до 582 дней).
Наиболее частыми нежелательными реакциями при лечении препаратом Ирелми, наблюдавшимися у ≥5% пациентов, являлись рвота и снижение концентрации витамина А.
Резюме нежелательных реакций
Нежелательные реакции указаны в соответствии с системно-органными классами Медицинского словаря для нормативно-правовой деятельности (MedDRA). В пределах каждого системно-органного класса термины предпочтительного употребления расположены в порядке уменьшения частоты, а затем в порядке уменьшения серьезности. Частота возникновения нежелательных реакций определяется как: очень часто (≥1/10), часто (≥1/100, но <1/10), нечасто (≥1/1000, но <1/100), редко (≥1/10000, но <1/1000), очень редко (<1/10000), частота неизвестна (на основании имеющихся данных оценить невозможно).
Таблица 4. Краткий обзор нежелательных реакций по категориям частоты
| Системно-органный класс | Нежелательная реакция | Частота |
| Со стороны органа зрения | Затуманивание зрения | Часто |
| Катаракта | Часто | |
| Со стороны сердца | Атриовентрикулярная блокада | Часто |
| Со стороны ЖКТ | Рвота | Часто |
| Общие нарушения и реакции в месте введения | Эритема в месте инъекции | Часто |
| Боль в месте инъекции | Часто | |
| Зуд в месте инъекции | Часто | |
| Лабораторные и инструментальные данные | Снижение концентрации витамина А | Очень |
| Протеинурия | Часто |
Описание отдельных нежелательных реакций
Снижение концентрации витамина А
В клиническом исследовании с участием пациентов с ATTRv-PN всех пациентов проинструктировали о необходимости приема рекомендуемой суточной нормы витамина А. У всех пациентов, получавших терапию препаратом Ирелми, исходная концентрация витамина А была нормальной, у 96.5% из них в ходе исследования концентрация витамина А стала ниже нижней границы нормы (см. раздел "Фармакологическое действие").
Реакции в месте инъекции
У пациентов с ATTRv-PN, получавших терапию препаратом Ирелми, эритема в месте инъекции, боль в месте инъекции и зуд в месте инъекции были отмечены в 3.5%, 3.5% и 2.1% случаев соответственно.
Сообщение о подозреваемых нежелательных реакциях
Важно сообщать о подозреваемых нежелательных реакциях после регистрации препарата с целью обеспечения непрерывного мониторинга соотношения "польза-риск" лекарственного препарата. Медицинским работникам рекомендуется сообщать о любых подозреваемых нежелательных реакциях лекарственного препарата через национальную систему сообщения о нежелательных реакциях государств-членов Евразийского экономического союза.
Противопоказания к применению
- повышенная чувствительность к эплонтерсену или к любому из вспомогательных веществ, входящих в состав препарата.
Применение при беременности и кормлении грудью
Женщины c детородным потенциалом
Применение препарата Ирелми снижает концентрацию витамина А в плазме крови, который необходим для нормального развития плода. Неизвестно, будет ли прием витамина А достаточным для снижения риска для плода. Поэтому до начала терапии препаратом Ирелми необходимо исключить беременность, а женщинам с детородным потенциалом следует использовать эффективные методы контрацепции.
Если женщина планирует беременность, применение препарата Ирелми и прием витамина А следует прекратить, а перед попыткой зачатия необходимо проконтролировать уровень витамина А в сыворотке крови, чтобы удостовериться в его нормализации. Из-за длительного периода полувыведения эплонтерсена (см. раздел "Фармакокинетика") дефицит витамина А может развиться даже после прекращения лечения.
Контрацепция
Женщинам с детородным потенциалом следует использовать эффективные методы контрацепции.
Беременность
Данные по применению препарата Ирелми у беременных женщин отсутствуют.
Введение эплонтерсена или фармакологически активного суррогата грызунам в дозах, в 38 раз превышающих рекомендуемую дозу для человека, в комбинированном исследовании репродуктивной токсичности и эмбриофетального развития не привело к влиянию на фертильность самцов и самок или развитие эмбриона и плода у мышей (см. подраздел "Данные доклинической безопасности").
В связи с потенциальным тератогенным риском, обусловленным несбалансированной концентрацией витамина А, препарат Ирелми не следует применять во время беременности. В случае беременности необходимо проводить тщательное наблюдение за состоянием плода и концентрацией витамина А, особенно в первом триместре.
Исходный риск серьезных врожденных дефектов развития плода и невынашивания беременности в целевой популяции неизвестен.
Период грудного вскармливания
Исследования периода лактации у человека или животных для оценки присутствия эплонтерсена или его метаболитов в грудном молоке, влияния на детей, находящихся на грудном вскармливании, или влияния на выработку молока у матери не проводились. Нельзя исключать риск для детей, находящихся на грудном вскармливании. Следует принять решение либо о прекращении грудного вскармливания, либо о приостановке/прекращении терапии препаратом Ирелми с учетом пользы грудного вскармливания для ребенка и пользы терапии для женщины.
Фертильность
Информация о влиянии эплонтерсена на фертильность человека отсутствует.
При введении эплонтерсена или фармакологически активного суррогата грызунам в дозах, обеспечивающих до 38 раз более высокую экспозицию, чем рекомендованная для человека, не выявлено влияния эплонтерсена на фертильность самцов или самок мышей.
Применение при нарушениях функции печени
Применение при нарушениях функции почек
Применение у детей
Применение у пожилых пациентов
Особые указания
Снижение уровня витамина А в сыворотке крови и прием витамина А
На основании механизма действия ожидается, что препарат Ирелми снижает концентрацию витамина А (ретинола) в сыворотке крови ниже нормальных значений (см. раздел "Фармакологическое действие").
Перед началом лечения препаратом Ирелми следует оценить любые симптомы или признаки, связанные с дефицитом витамина А.
Пациентам, получающим препарат Ирелми, следует принимать витамин А в рекомендуемой суточной дозе для уменьшения потенциального риска развития симптомов со стороны органа зрения, обусловленных дефицитом витамина А. Не следует назначать более высокие дозы витамина А, свыше рекомендуемых суточных доз, для достижения нормального уровня витамина А в сыворотке крови во время лечения препаратом Ирелми, поскольку уровень витамина А в сыворотке крови не отражает общего содержания витамина А в организме.
При появлении симптомов со стороны органа зрения, соответствующих дефициту витамина А, включая ухудшение ночного зрения или ночную слепоту, а также постоянную сухость глаз, следует провести офтальмологическое обследование.
Неизвестно, будет ли прием витамина А во время беременности достаточным для профилактики дефицита витамина А, если беременная женщина продолжает получать препарат Ирелми (см. раздел "Беременность и лактация"). Увеличение дозы витамина А сверх рекомендуемой во время беременности вряд ли приведет к коррекции концентрации ретинола в сыворотке крови в связи с механизмом действия эплонтерсена и может быть вредным для матери и плода.
Вспомогательные вещества
Содержание натрия
Данный препарат содержит менее 1 ммоль (23 мг) натрия на 0.8 мл, т.е., по сути, не содержит натрия.
Влияние на способность к управлению транспортными средствами и механизмами
Препарат Ирелми не оказывает влияния или оказывает незначительное влияние на способность управлять транспортными средствами и работать с механизмами.
Передозировка
Симптомы: специфические симптомы передозировки эплонтерсена неизвестны.
Лечение: специфическое лечение при передозировке эплонтерсена отсутствует. В случае передозировки необходимо проконсультироваться с врачом и проводить симптоматическую терапию.
Лекарственное взаимодействие
Формальные исследования лекарственных взаимодействий не проводились (см. раздел "Фармакокинетика").
В связи с отсутствием исследований совместимости данный препарат не следует смешивать с другими лекарственными препаратами.
Условия хранения препарата Ирелми
Препарат следует хранить при температуре от 2° до 8°C в оригинальной упаковке. Не замораживать.
Препарат Ирелми можно хранить в оригинальной картонной упаковке при комнатной температуре ниже 30°C не более шести недель. Если препарат не был использован в течение шести недель, его следует утилизировать.
Условия реализации
Препарат отпускают по рецепту.
Контакты для обращений
АСТРАЗЕНЕКА ФАРМАСЬЮТИКАЛЗ ООО (Россия)

|
|
123112 Москва, |
 --%3E%3Csvg version='1.1' id='Слой_1' xmlns='http://www.w3.org/2000/svg' xmlns:xlink='http://www.w3.org/1999/xlink' x='0px' y='0px' viewBox='0 0 14.17 14.17' style='enable-background:new 0 0 14.17 14.17;' xml:space='preserve'%3E%3Cstyle type='text/css'%3E .st0%7Bfill:%23B7013A;%7D%0A%3C/style%3E%3Cg%3E%3Cpath class='st0' d='M7.72,11.51c-4.84,0-7.6-3.32-7.72-8.84h2.43c0.08,4.05,1.87,5.77,3.28,6.13V2.66h2.28v3.5 c1.4-0.15,2.87-1.74,3.36-3.5h2.28c-0.38,2.16-1.97,3.75-3.11,4.41c1.13,0.53,2.95,1.92,3.64,4.44h-2.51 c-0.54-1.68-1.89-2.98-3.66-3.16v3.16H7.72z'/%3E%3C/g%3E%3C/svg%3E%0A)
 --%3E%3Csvg version='1.1' id='Слой_1' xmlns='http://www.w3.org/2000/svg' xmlns:xlink='http://www.w3.org/1999/xlink' x='0px' y='0px' viewBox='0 0 14.17 14.17' style='enable-background:new 0 0 14.17 14.17;' xml:space='preserve'%3E%3Cstyle type='text/css'%3E .st0%7Bfill:%23B7013A;%7D%0A%3C/style%3E%3Cg%3E%3Cg%3E%3Cpath class='st0' d='M7.15,0C5.12,0,3.47,1.64,3.47,3.67c0,2.03,1.64,3.67,3.67,3.67s3.67-1.64,3.67-3.67 C10.82,1.64,9.17,0,7.15,0z M7.15,5.19c-0.84,0-1.52-0.68-1.52-1.52s0.68-1.52,1.52-1.52s1.52,0.68,1.52,1.52S7.98,5.19,7.15,5.19 z'/%3E%3C/g%3E%3Cg%3E%3Cg%3E%3Cpath class='st0' d='M8.44,10.27c1.47-0.3,2.35-1,2.4-1.03c0.43-0.35,0.5-0.97,0.15-1.4c-0.35-0.43-0.97-0.5-1.4-0.15 C9.58,7.68,8.64,8.4,7.1,8.41c-1.55,0-2.51-0.72-2.51-0.73C4.15,7.33,3.52,7.4,3.18,7.83C2.83,8.26,2.9,8.89,3.33,9.23 c0.05,0.04,0.96,0.75,2.48,1.04l-2.11,2.2c-0.38,0.4-0.37,1.03,0.03,1.41c0.19,0.19,0.44,0.28,0.69,0.28 c0.26,0,0.52-0.1,0.72-0.31l1.96-2.08l2.15,2.1c0.39,0.39,1.02,0.39,1.41,0c0.39-0.39,0.39-1.02,0-1.41L8.44,10.27z'/%3E%3C/g%3E%3Cg%3E%3Cpath class='st0' d='M7.1,8.41C7.09,8.41,7.1,8.41,7.1,8.41C7.09,8.41,7.1,8.41,7.1,8.41z'/%3E%3C/g%3E%3C/g%3E%3C/g%3E%3C/svg%3E%0A)
 --%3E%3Csvg version='1.1' id='Слой_1' xmlns='http://www.w3.org/2000/svg' xmlns:xlink='http://www.w3.org/1999/xlink' x='0px' y='0px' viewBox='0 0 14.17 14.17' style='enable-background:new 0 0 14.17 14.17;' xml:space='preserve'%3E%3Cstyle type='text/css'%3E .st0%7Bfill-rule:evenodd;clip-rule:evenodd;fill:%23B7013A;%7D%0A%3C/style%3E%3Cg%3E%3Cpath class='st0' d='M0.97,6.28C4.77,4.63,7.31,3.53,8.58,3c3.62-1.51,4.38-1.77,4.87-1.78c0.11,0,0.35,0.02,0.51,0.15 c0.13,0.11,0.16,0.25,0.18,0.36c0.02,0.11,0.04,0.34,0.02,0.51c-0.2,2.06-1.04,7.08-1.48,9.38c-0.18,0.98-0.54,1.3-0.89,1.34 c-0.76,0.07-1.34-0.5-2.06-0.98c-1.15-0.75-1.79-1.22-2.91-1.96C5.52,9.17,6.36,8.7,7.1,7.94c0.2-0.2,3.53-3.24,3.6-3.51 c0.01-0.03,0.01-0.16-0.07-0.23c-0.08-0.07-0.18-0.04-0.27-0.02c-0.12,0.02-1.95,1.24-5.5,3.64C4.34,8.18,3.87,8.36,3.45,8.34 C2.98,8.33,2.09,8.08,1.42,7.87C0.6,7.61-0.05,7.46,0,7.01C0.04,6.77,0.36,6.53,0.97,6.28z'/%3E%3C/g%3E%3C/svg%3E%0A)
 --%3E%3Csvg version='1.1' id='Слой_1' xmlns='http://www.w3.org/2000/svg' xmlns:xlink='http://www.w3.org/1999/xlink' x='0px' y='0px' viewBox='0 0 14.17 14.17' style='enable-background:new 0 0 14.17 14.17;' xml:space='preserve'%3E%3Cstyle type='text/css'%3E .st0%7Bfill:%23B7013A;%7D%0A%3C/style%3E%3Cg%3E%3Cpath class='st0' d='M7.53,0.91C7.53,0.91,7.53,0.91,7.53,0.91L7.53,0.91c-0.94,0-1.82,0.19-2.64,0.55L3.44,0 C1.9,0.68,0.67,1.91,0,3.46l1.46,1.46c0.67-1.55,1.9-2.78,3.44-3.46l5.28,5.28c0.04,0.02,0.08,0.05,0.1,0.1l0.02,0.02 c0,0,0,0-0.01,0c0,0.01,0.01,0.02,0.01,0.03l0.38,1.81c0,0,0,0,0,0l0.52,2.53l-2.52-0.52c0,0,0,0,0,0l-1.8-0.39 c-0.01,0-0.02-0.01-0.02-0.01c0,0,0,0.01-0.01,0.01l-0.03-0.03c-0.03-0.02-0.06-0.05-0.08-0.08L1.46,4.91 C1.09,5.74,0.9,6.64,0.9,7.55c0,3.66,2.98,6.63,6.64,6.63c3.66,0,6.63-2.97,6.63-6.64C14.17,3.88,11.2,0.91,7.53,0.91z'/%3E%3Cpath class='st0' d='M8.88,10.31c0.34-0.6,0.83-1.09,1.43-1.44c-0.01-0.01-0.01-0.02-0.02-0.04l-0.38-1.8 C8.68,7.67,7.68,8.68,7.05,9.91l1.79,0.39C8.86,10.3,8.87,10.31,8.88,10.31z'/%3E%3C/g%3E%3C/svg%3E%0A)
 --%3E%3Csvg version='1.1' id='Слой_1' xmlns='http://www.w3.org/2000/svg' xmlns:xlink='http://www.w3.org/1999/xlink' x='0px' y='0px' viewBox='0 0 14.17 14.17' style='enable-background:new 0 0 14.17 14.17;' xml:space='preserve'%3E%3Cstyle type='text/css'%3E .st0%7Bfill:%23B7013A;%7D%0A%3C/style%3E%3Cg%3E%3Cpath class='st0' d='M3.11,11.34V10.8H2.17c-0.67,0-1.21-0.55-1.21-1.21V2.17c0-0.67,0.55-1.21,1.21-1.21h5.75 c0.4,0,0.75,0.2,0.97,0.5h1.07C9.66,0.61,8.86,0,7.92,0H2.17C0.97,0,0,0.98,0,2.17v7.42c0,1.2,0.97,2.17,2.17,2.17h0.98 C3.13,11.63,3.11,11.48,3.11,11.34z'/%3E%3Cg%3E%3Cpath class='st0' d='M8.73,9.07c-0.09,0-0.18-0.04-0.25-0.11C7.88,8.37,7.86,7.44,8.41,6.89l1.38-1.38 c0.27-0.27,0.64-0.42,1.04-0.4c0.39,0.01,0.75,0.18,1.04,0.46c0.28,0.28,0.44,0.65,0.46,1.04c0.02,0.4-0.13,0.77-0.4,1.04 l-0.68,0.68c-0.14,0.14-0.37,0.14-0.51,0c-0.14-0.14-0.14-0.37,0-0.51l0.68-0.68c0.13-0.13,0.2-0.31,0.19-0.5 c-0.01-0.2-0.1-0.4-0.25-0.55s-0.35-0.24-0.55-0.25c-0.19-0.01-0.37,0.06-0.5,0.19L8.92,7.4C8.65,7.67,8.67,8.14,8.98,8.45 c0.14,0.14,0.14,0.37,0,0.51C8.91,9.03,8.82,9.07,8.73,9.07z'/%3E%3Cpath class='st0' d='M7.48,11.48c-0.39,0-0.79-0.15-1.09-0.46C5.8,10.43,5.77,9.5,6.33,8.94l0.68-0.68c0.14-0.14,0.37-0.14,0.51,0 c0.14,0.14,0.14,0.37,0,0.51L6.84,9.45C6.57,9.72,6.59,10.2,6.9,10.51c0.31,0.31,0.78,0.33,1.06,0.06l1.38-1.38 c0.27-0.28,0.25-0.75-0.06-1.06c-0.14-0.14-0.14-0.37,0-0.51c0.14-0.14,0.37-0.14,0.51,0c0.59,0.59,0.61,1.52,0.06,2.08 l-1.38,1.38C8.2,11.34,7.84,11.48,7.48,11.48z'/%3E%3C/g%3E%3Cpath class='st0' d='M12,2.41H6.25c-1.2,0-2.17,0.98-2.17,2.17V12c0,1.2,0.97,2.17,2.17,2.17H12c1.2,0,2.17-0.98,2.17-2.17V4.58 C14.17,3.38,13.2,2.41,12,2.41z M13.21,12c0,0.67-0.55,1.21-1.21,1.21H6.25c-0.67,0-1.21-0.55-1.21-1.21V4.58 c0-0.67,0.55-1.21,1.21-1.21H12c0.67,0,1.21,0.55,1.21,1.21V12z'/%3E%3C/g%3E%3C/svg%3E%0A)
X